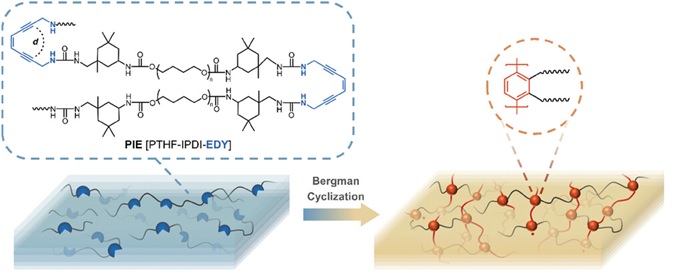
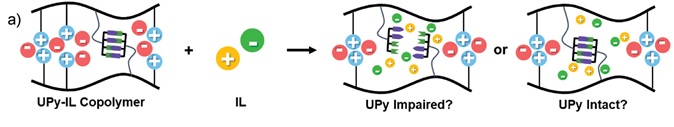
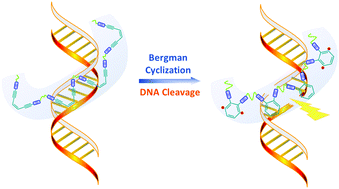
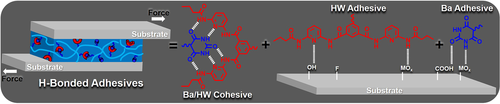
Gloger, D. et. al. ACS Applied Polymer Materials, 2024, 6, 10824-10841, https://doi.org/10.1021/acsapm.4c01938
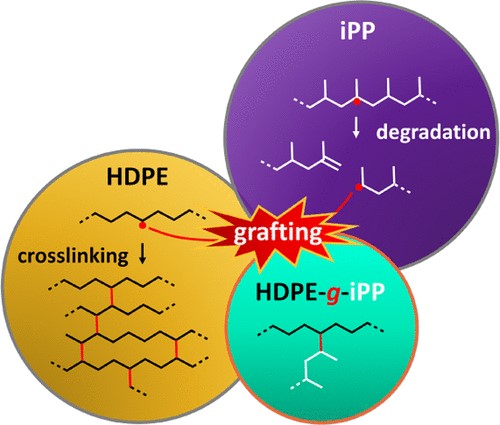
In this study, we report the coupling of high-density polyethylene (HDPE) and isotactic polypropylene (iPP) into grafted block copolymers (HDPE-g-iPP) from HDPE and iPP precursors subjected to reactive extrusion with an organic peroxide, however, without a cross-linker. Coupling of macroradicals in the melt state is confined to a small interfacial volume at the HDPE/iPP domain interfaces of this immiscible two-phase blend system. The tendency of HDPE macroradicals to branch and cross-link into a solid-like network, together with the tendency of iPP macroradicals to simply cleave, is a common problem to overcome. Moreover, to prove HDPE-g-iPP molecules in reactive HDPE/iPP blends is difficult due to the low grafting yields and the additional challenge to analytically distinguish HDPE-g-iPP molecules from the HDPE/iPP matrix. In this study, we work with a low-viscosity blend system, which, combined with the low unsaturation content of the HDPE, made cross-linking negligible. We identify HDPE-g-iPP molecules via interaction chromatography by iPP components in the HDPE elution range and by analytical temperature-rising elution fractionation, where we find HDPE in the iPP elution range. Physical characterization by thermal, dynamic mechanical, and morphological analyses confirmed that the reactive blend is compatibilized by HDPE-g-iPP molecules, seen by shifts in crystallization temperature and glass transition, and by diffuse domain interfaces. With improved grafting yields, reactive blends could potentially be used directly as compatibilizers. Copyright © 2024, American Chemical Society.